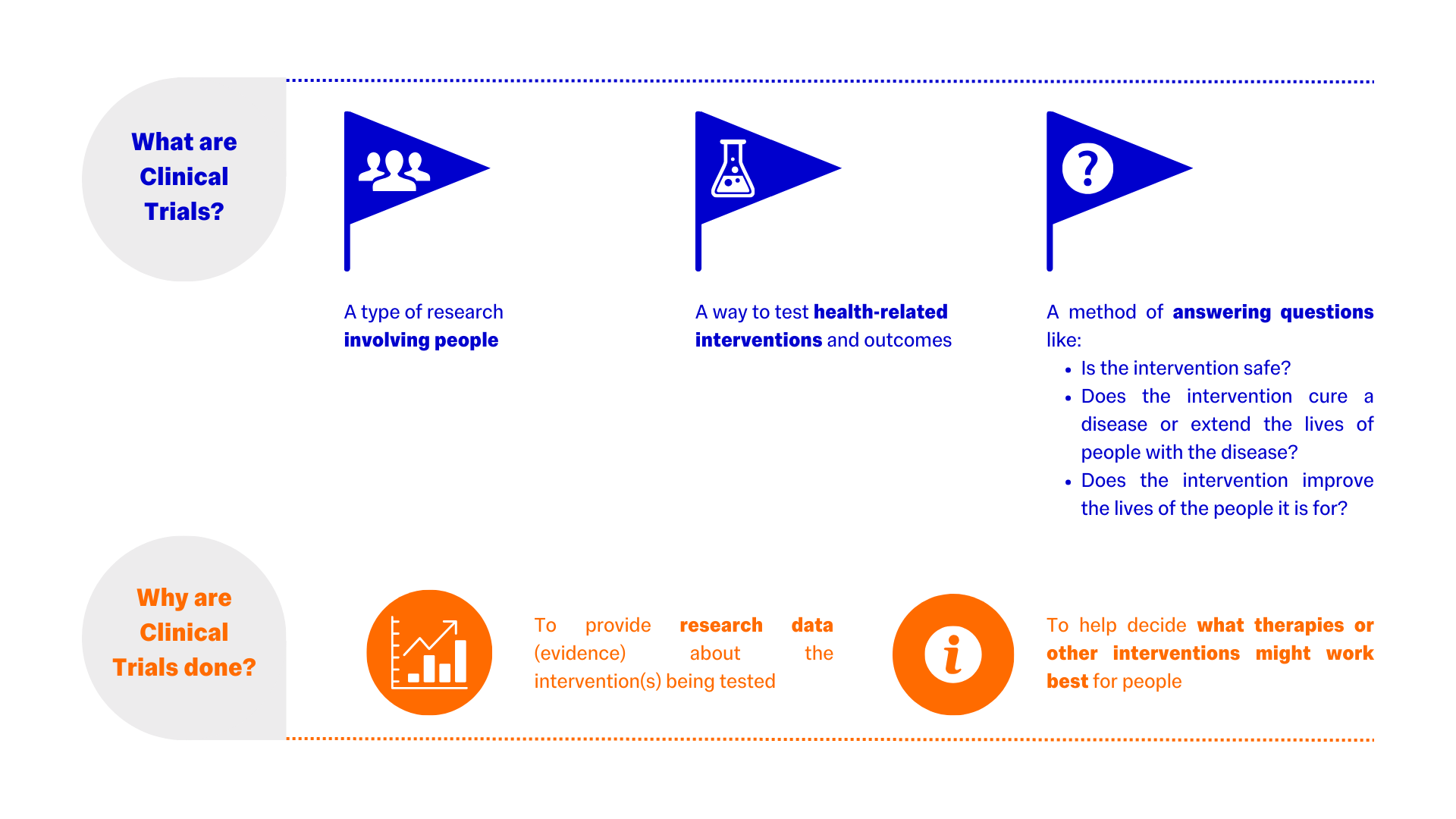
What are Clinical Trials?

Types of Clinical Trial
- Advanced Therapy Medicinal Trial (ATMP) or Advanced Therapy Investigational Medicinal Trial (ATIMP)
- Clinical Trial of an Investigational Medicinal Product (CTIMP)
- Medical Device Clinical Trial (MDCT)
- non-CTIMP Studies
ATMP - As defined by Article 2 (1) of Regulation 1394/2007, a medicinal product for human use that is either a gene therapy medicinal product, a somatic cell therapy medicinal product, or a tissue engineered product.
ATIMP - An ATMP as defined above that is used within a clinical trial in accordance with Article 2(d) of Directive 2001/20/EC.
A clinical trial within the scope of the UK Medicines for Human Use (Clinical Trials) Regulations 2004.
An investigation in human subjects intended:
a) to discover or verify the clinical, pharmacological and/or other pharmacodynamic effects of one or more medicinal products,
b) to identify any adverse reactions, and/or
c) to study absorption, distribution, metabolism and excretion, with the object of ascertaining the safety and/or efficacy of those products.
Investigations undertaken to assess or test the safety and efficacy of a given medical device or innovation. These projects usually test the devices’ performance in preventing, diagnosing, or treating a particular condition.
Trials that do not involve an investigational medicinal product and therefore do not fall under the UK Medicines for Human Use (Clinical Trials) Regulations 2004.
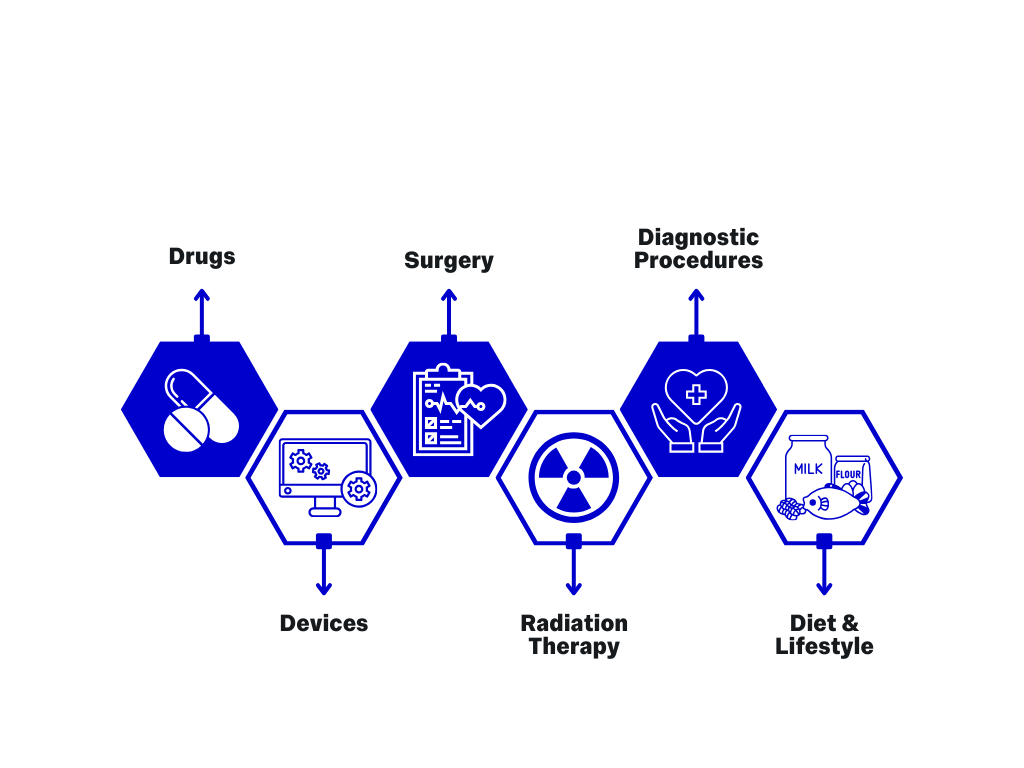
Health Interventions Studied in Clinical Trials
How can this be applied to Medical Technologies?
Clinical trials can:
- Test the efficacy and functioning in practice of novel medical technologies
- Test novel medical technologies against the benchmark of existing technologies within the field
- Demonstrate the benefits of novel technologies, or redesigns of existing technologies when compared to existing and adopted technologies
- Give scientific backing to novel technologies and begin an evidence-base that could lead to adoption within the wider field
Ethics: Do I need to apply?
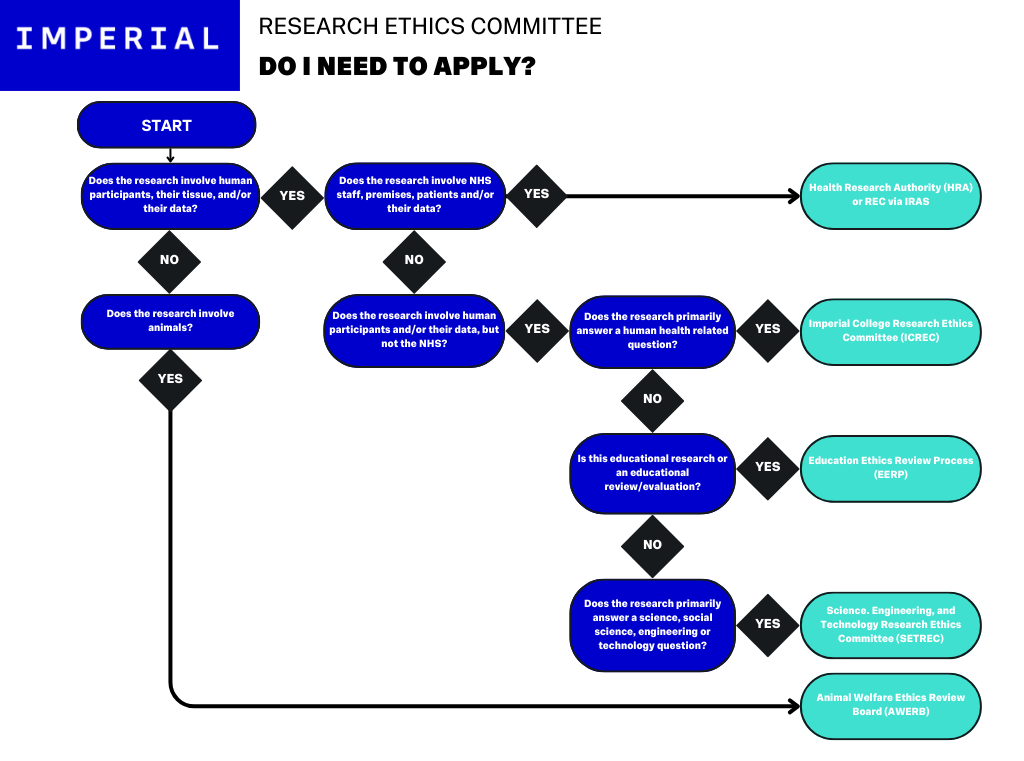
The above flow chart demonstrates the decision tree for the different Ethics Committees that may apply to research conducted at Imperial. If you are uncertain as to which REC to apply to, check with your Chief Investigator.
For a more detailed look at Research Ethics, see the dedicated Research Ethics at Imperial Resource.
Clinical Trial Requirements
To be considered a clinical trial, a study must:
- Be conducted on human subjects – no study in non-human animals is considered a clinical trial.
- Investigate an intervention – the study treats or diagnoses its subjects with a drug, medical device, gene-therapy or other medical intervention. If your study is not an interventional trial, then it is likely an observational study (E.G.: a case-control, cross-sectional, or cohort study).
- Look for an effect – the study must collect data on measures chosen during trial design.
- Compare the intervention to a control – a portion of subjects are given no treatment, a placebo, or the best available treatment other than the intervention. The effects for both the intervention and control group are compared.
- Be prospective – subjects must be studied forward in time, which means retrospective studies are not classed as clinical trials.
The MHRA has designed an algorithm to help researchers check if their project is classed as a clinical trial. You can access it here.
Clinical Trials - Trial Startup
Overview
Trial set up processes can vary between CTIMPs, non-CTIMPs and other trial types. The following advice is general, you should speak to your relevant departmental expert for more tailored guidance.
You should consider:
- The Research Question - The nature of the question will determine which regulations the trial must adhere to.
- Where are you in the trial process? - The key stages are shown in the pathway on the next page.
- Organisation and trial team expertise - Use the following pathway to identify any skills gaps in your team, and see the information on sourcing expertise at Imperial in the following pages.
- The importance of Patient & Public Involvement - Consider how best to involve members of the public in your work including using Imperial’s PPIE Team.
Clinical Trials Pathway
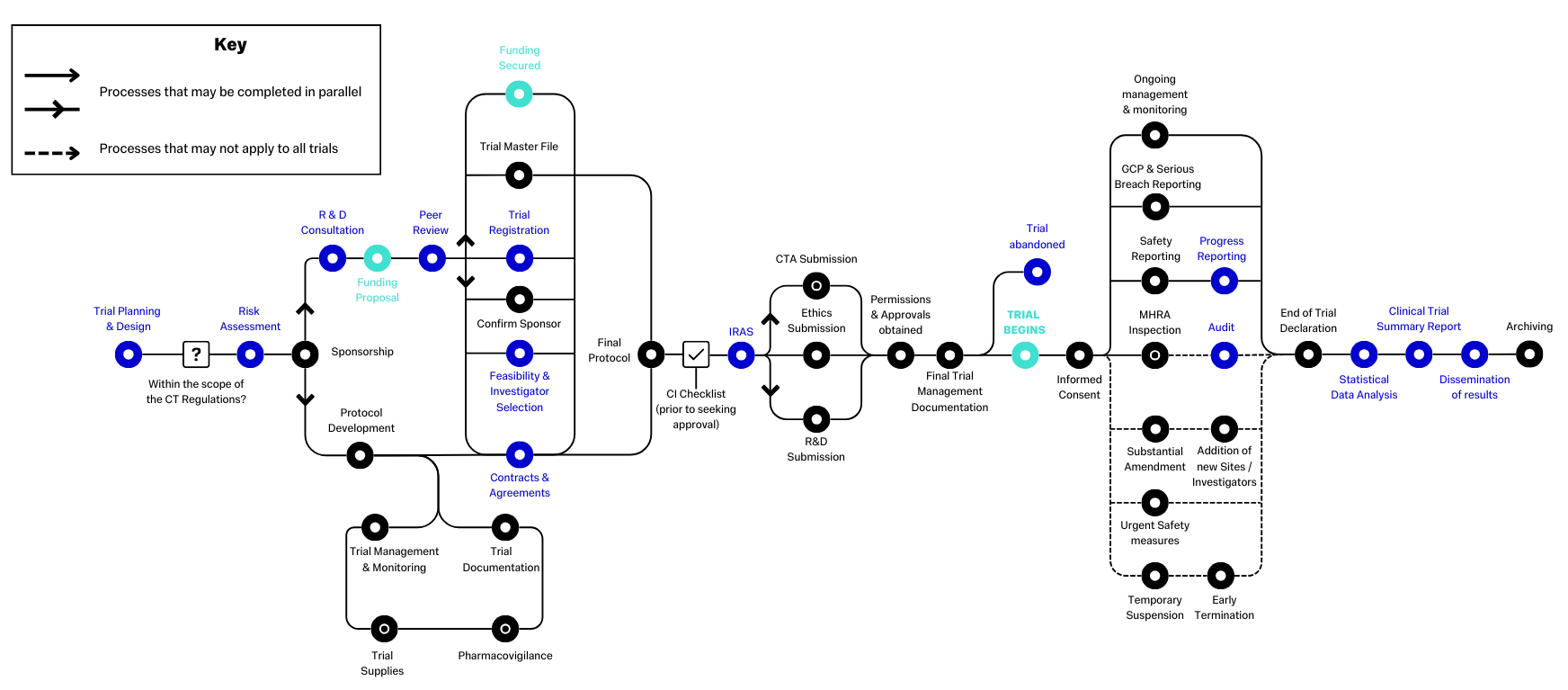
This diagram demonstrates the standard pathway that clinical trials follow. Items in black and green are standard items that will follow a set process, whereas items in blue may not apply to all studies, or may follow study specific processes. An example of this is "Contracts & Agreements" - The types of contracts, or other agreements, you need will vary depending on the nature of the project.
Created by the NIHR, the original diagram and surrounding information are available at Routemap (ct-toolkit.ac.uk)
Trial Design & Planning
Good study design is important.
The trial design should be considered before the protocol is developed to ensure that all requirements are identified and met, particularly with regard to appropriate funding sources and amounts,
A good study plan will help with the development of funding applications, ethics committee and R&D approvals, NHS approvals, and any other regulatory permissions required.
Imperial offers a research design service that can support the development of your study design, find out more here.
Important Study Documents
- Delegation Log
- Funding Agreements
- Investigator Site File (ISF)
- Record of REC Approval (Favourable Opinion)
- Research Agreements / Contracts
- Trial Master File (TMF)
- Trial Team: CVs
- Trial Team: Insurance
- Trial Team: Training & Certifications
The Delegation Log is a vital record of the project or study team and designates the responsibilities and permissions of the team for the execution of the project.
The Delegation Log is a good example of a living document. It should be checked regularly for accuracy and should be updated promptly when there are any changes to the project team or their responsibilities.
The specific documents recording any funding you are being awarded, the financial amounts, from whom the funding is being provided, and the duration for which the funding is valid. These documents will also cover any stipulations or conditions relating to the funding, such as the items or project needs the funding may be used for.
The ISF includes all of the important documentation that demonstrates the project is being undertaken in line with regulatory requirements.
This includes items like the SOP, participant consent forms and information sheets, delegation logs, safety reports, IMP manuals etc.
ISFs should be kept at all sites.
The official letter of the Research Ethics Committee’s approval of your project, also known as a “Favourable Opinion”.
You must have this in place before starting your project.
Written records of agreements with your Sponsor, the research facility or facilities where your project will take place, and any other similar agreements. Some projects will have dissemination agreements which lay out how the results of the project will be made public after the end of the study.
Similar to the ISF, the TMF forms a record of the important project documents, but is usually kept solely at the coordinating project site, under the supervision of the CI or study leads.
The TMF should contain all major documents related to the project, from funding agreements and contracts, to copies of the SOP and participant-facing documents, to the statistical analysis plan.
You must obtain and store the CVs of all team members who will be working on the project so that you have a written record demonstrating the competencies and qualifications of your team.
Some team members, usually senior team such as the CI / PI, or clinical staff like doctors and nurse, may require liability insurance to be in place when they work on your study. For clinical staff, this may be taken care of by the NHS or their normal employer.
You may also need more general liability or indemnity insurance for your project - Check with your Sponsor.
You should store copies of any training certificates obtained by, or required by, your team. This can include Good Clinical Practice training, First Aid, ANTT training, and data security trainings, amongst others.
Other Study Documents
- Case Report Form (CRF / eCRF)
- Consent Form
- Other information and templates
- Participant Information Sheet (PIS)
- Participant Information Sheet Summary
- Standard Operating Procedures (SOPs) / Manual of Procedures (MOPs)
The CRF or eCRF record all data related to the project. They may be completed at participant appointments, and will be the primary data source for the project. The CRF should be thorough, and should be designed to record all relevant data.
This form will be read, understood, and signed by participants after they have read the PIS and have had time to ask questions about the project. The consent form should thoroughly record consent for all aspects of participation in the project.
Imperial-approved templates and further information on study documents can be found here.
This is the document that will form the basis of the information prospective participants receive about the project. It should be detailed enough for participants to make an informed choice about joining the project, but should be in clear and easy-to-understand language.
Provided to participants, this document should succinctly summarise the PIS, which may be quite lengthy depending on the project, into a single side of A4. You should try to include all the key information, and should make an effort not to omit any major points.
This document includes the full information about the project. It should include a brief summary of the supporting literature, but should primarily focus on the subject of the project, how you plan to carry out the project, and specific items such as participant recruitment criteria, appointment structures, and outputs.
Sponsor
The Sponsor is responsible for ensuring appropriate arrangements are in place for:
- The Site Initiation Visit (SIV)
- The management and oversight of the project.
- The overall management of financing the project.
More guidance on the process at Imperial can be found here.
Site Initiation Visit

Once all of the relevant approvals are in place, all documentation has been finalised, and all sites have the information they need, the trial can go ahead.
Typically, a site initiation meeting, or “visit”, is held at each site (in person or virtual - N.B. The core research team must attend in person where possible). The Chief Investigator is then able to confirm all technical aspects of a trial and that protocol requirements are fully understood by all site staff, and necessary documents are in place.
The visit also provides the site team with an opportunity to ask questions. Trial or facility specific training normally happens at this stage.
Resources at Imperial
Facilities at Imperial
If you are an Imperial researcher or staff member, or if your project is Sponsored by Imperial, you may be eligible to access the Imperial NHS Trust Clinical Research Facility (ICRF). The ICRF is a clinical facility at Hammersmith Hospital, London, specifically designed to accommodate research projects.
You can find information on the ICRF here.
Spinouts at Imperial
Imperial is committed to supporting spinouts for Imperial-developed innovations. The point at which you spinout will depend on the specific innovation, and any specific project needs.
You can find further guidance on spinouts at Imperial here.
Resource Download / Contact Us
You can download a copy of these resources here: Clinical Trial Basics (PDF) and here: Clinical Trial Start Up (PDF) . These PDFs include additional information, such as suggested reading lists, glossaries, and contact information.
If you would like to contact us, or if there is a resource you would like to see, reach out to us using this form!
