
Collaborative Resource: Research Ethics at Imperial
Summary

All projects and studies involving people as participants
All projects and studies involving people as participants will need to be reviewed by a Research Ethics Committee (REC).

REC review is the responsibility of the Chief Investigator.
REC review is the responsibility of the Chief Investigator (or Principal Investigator, or lead researcher on the study).

RECs consider the safety and ethics of the proposed study.
RECs consider the safety and ethics of the proposed intervention or study design, fairness to the study population, burden to participants.

REC review is usually conducted by a University or NHS REC
In the UK, ethical review is usually sought from the lead researcher’s university or from one of the Health Research Authority’s (HRA) NHS RECs.

You may also need a sponsor. Your research office can help.
You may also need a sponsor. If you are a researcher based in a university or the NHS, your local research office can offer you guidance.
Understanding the Different Research Ethics Committees
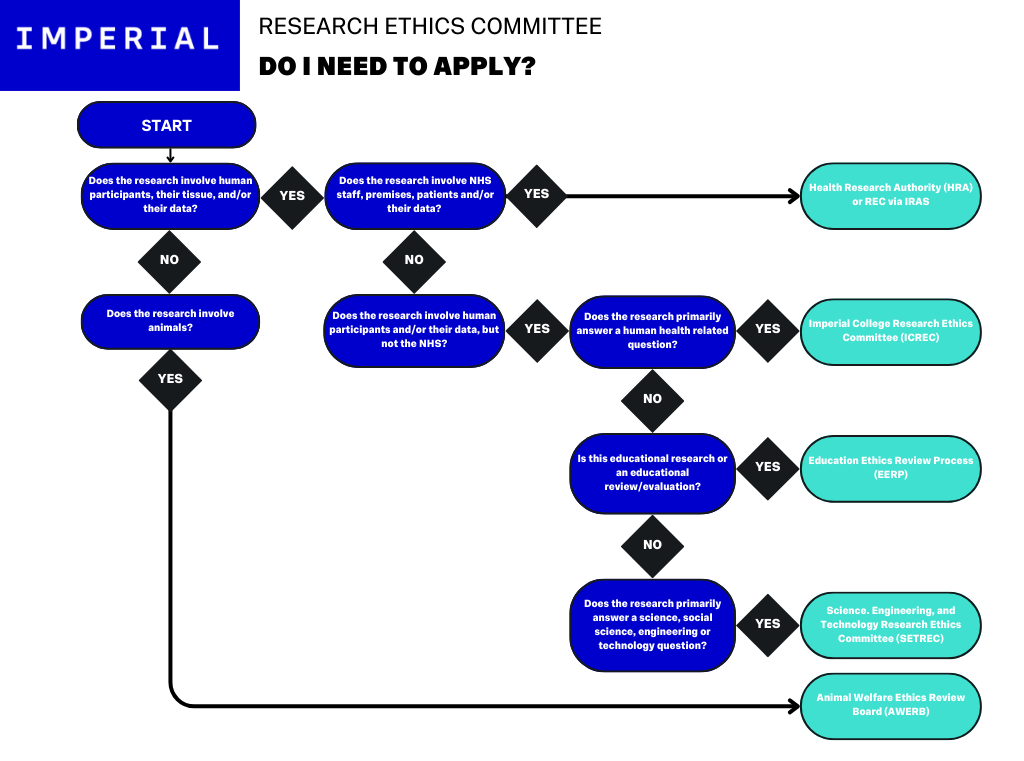
The above image demonstrates the decision tree for the different Ethics Committees that may apply to research conducted at Imperial. If you are uncertain as to which REC to apply to, check with your Chief Investigator.
Research Ethics Committees: An overview
- The Animal Welfare Ethics Review Board (AWERB)
- The Educational Ethics Review Process (EERP)
- Imperial College Research Ethics Committee (ICREC)
- The Science, Engineering, and Technology Research Ethics Committee (SETREC)
- The Health Research Authority (HRA), Medicines and Healthcare Research Authority (MHRA), and other RECs through the IRAS
Regulated by the Animals (Scientific Procedures) Act 1986 and governed by the Home Office (Secretary of State), the AWERB ensures that any proposed work involving animal subjects is well justified and is carried out to the highest ethical standards.
It is recommended that you read the Animals (Scientific Procedures) Act 1986 to best understand both the legal requirements of animal work, and your responsibilities as a researcher.
Visit the Imperial Animals in Research website for further information, or alternatively the gov.uk site “Animal testing and research: Guidance for the regulated community”.
The EERP is designed only for educational projects, such as undergraduate dissertation projects. High-risk projects may be forwarded to SETREC (below) for further review; you will be notified if this happens.
Visit the EERP at Imperial website for further information as well as an email contact for queries.
The dedicated REC for projects conducted by Imperial College staff or students, where the project is directly health-related and uses human participants and / or their data.
Visit the ICREC website for more information on Committee meeting timetables and submission deadlines.
A dedicated review board for non-health related research being conducted by Imperial staff or students, using human participants and / or their data. This committee also reviews high-risk educational projects that cannot be approved via the EERP.
Visit the SETREC at Imperial website for further information, including meeting dates and submission deadlines.
Applications submitted via the Integrated Research Application System (IRAS) will be automatically directed to either the HRA or MHRA as needed for an assessment of the governance, regulatory, and legal compliance of the proposed project, as well as to a research ethics committee for an independent ethical review of the project. (Note that this only applies to the NHS in England and Wales, projects for locations in Scotland or Northern Ireland will need to have applications submitted via the appropriate system for the lead country).
The IRAS has the benefit of being an all-in-one system. You submit one application and will receive (where applicable) one “Request for Further Information (RFI)”, and once the committees are satisfied, you will receive one outcome, either a favourable or non-favourable opinion. The HRA or MHRA decision may be sent separately, so ensure you keep copies of all outcomes and communications in the Trial Master File.
HRA Approval is required for projects that are:
- Clinical Trials of an Investigational Medicinal Product (CTIMP) (with the exception of Phase 1 trials in healthy volunteers taking place outside the NHS);
- Clinical Investigations or other studies of a Medical Device;
- Combined trials of an Investigational Medicinal Product and an Investigational Medical Device
- Clinical Trials to study a novel intervention or randomised Clinical Trial to compare interventions in clinical practice;
- Basic science studies involving procedures with human participants;
- Studies administering questionnaires / interviews for quantitative analysis, or using mixed qualitative / quantitative methodology;
- Studies involving qualitative methods only;
- Studies limited to working with human tissue samples (or other human biological samples) and data (specific project only),
- Studies limited to working with data (specific project only).
If your project is not one of the above study types, but is one of the following, you will not need HRA approval but may still need REC approval:
- A Research Tissue Bank;
- A Research Database;
- Taking place in a non-NHS setting e.g. Phase 1 clinical trial in healthy volunteers or similar.
N.B. If your project involves accessing confidential patient information without consent, you must submit an application to the Confidentiality Advisory Group (CAG).
For further information, visit the HRA website.
The Integrated Research Application System: IRAS
The Integrated Research Application System (IRAS) is an online tool used to submit ethics applications for health and social care focused research, using human participants and / or their data. The system requires only one submission for a project, and will automatically handle any additional approvals required, such as HRA approval.
Completed in browser, the online system is comprised of the main submission form where you will fill in the information about your project, and an upload area where you can add your project documents (the main items of which are listed in the section below). You can save your submission at any point and return to it later. The system also allows you to electronically request signatures and approvals from key team members such as your Chief Investigator, and co-Investigators.
The “E-Learning” tab on the IRAS website contains guides and instructions for submitting a project via IRAS. This guide is very useful and can be found here.
- Project design complete, final drafts of Standard Operating Procedures / Manual of Procedures.
- Study document final drafts.
- Funding should be secured, with documented evidence.
- Sponsor should be in place, with documentation like Letters of Support.
- Main study team should be on board, particularly the Chief Investigator, Principal Investigator (where applicable), and study management.
PPIE should be in place, as you should have used these processes during the design phase. The Imperial PPIE team can provide support and guidance - Find out more here.
The following list of documents is a non-exhaustive list of documents and forms you should have prepared for your ethics submission. This could change depending on the review process you are applying to, so ensure you read all relevant provided guidance well ahead of your submission date to make sure you have not missed anything.
- Research proposal document; alternatively, this may be transcribed into forms such as IRAS.
- Budget and funding:
- Proposed budget
- Evidence of funding secured
- Letters of Support (from applicable institutions or Sites)
- Sponsor Agreement and documentation
- Standard Operating Procedures (SOP) / Manual of Procedures (MOP)
- Participant Information Sheet
- Participant Information Sheet Summary
- Consent Form (template)
- Expenses Form (template)
- Manuals, instructions, or specifications for any technology or medicinal product being studied.
- Study team:
- CVs
- Insurance documentation (where applicable)
- Certificates and training evidence (where applicable)
- Animal studies only: Proposed scheme of work involving animals.
Over time it may become apparent that you need to make a change to your project. This might be something like:
- Adding a new Site
- Changing a diagnostic tool being used
- Changing a process within the project
- Adding or removing team members
- Other, study-specific changes
Depending on what you are changing about your project this may need to be submitted to the REC as an Amendment.
Non-substantial Amendments refer to small changes that should not affect the outcomes of the project in any significant way. For example, adding a new Site is usually considered a non-substantial amendment as it does not change any of the project’s processes or execution.
Substantial Amendments refer to larger changes that may have an impact on the project outcomes, or how the project is being run. For example, if you discover a fault in how a piece of technology functions that may impact patient safety, you would need to fix the problem immediately, but because the project has already outlined the way the technology functions (and this is what was approved by the REC), you will likely need to submit an amendment to be approved by the REC.
For NHS-based research using human participants, amendments will be submitted by opening your project in the IRAS and completing the relevant amendment form. Read any instructions and guidance carefully. For other kinds of research, where possible, liaise with the REC or process that approved your project as they may have more specific guidance.
If in doubt, check in with your project supervisor, Sponsor, or Chief Investigator.
The Health Research Authority (HRA)
You will need HRA approval if your research project is:
- A Clinical Trial of an Investigational Medicinal Product (CTIMP) (with the exception of Phase 1 trials in healthy volunteers taking place outside the NHS).
- A Clinical Investigation or other study of a Medical Device.
- A combined trial of an Investigational Medicinal Product and an Investigational Medical Device.
- A Clinical Trial to study a novel intervention or randomised Clinical Trial to compare interventions in clinical practice.
- A basic science study involving procedures with human participants.
- A study administering questionnaires/interviews for quantitative analysis, or using mixed qualitative/quantitative methodology.
- A study involving qualitative methods only.
- A study limited to working with human tissue samples (or other human biological samples) and data (specific project only).
- A study limited to working with data (specific project only).
HRA applications can be handled automatically via the IRAS system.
If your project is:
- a Research Tissue Bank,
- a Research Database,
- taking place in a non-NHS setting,
you will not need HRA approval, but you may still require REC approval.
Principles that apply to all Health & Social Care Research
- A. Safety
- B. Competence
- C. Scientific & Ethical Conduct
- D. Patient, Service User, and Public Involvement
- E. Integrity, Quality and Transparency
- F. Protocol
- G. Legality
- H. Benefits and Risks
- I. Approval
- J. Information about the Research
- K. Accessible Findings
- L. Choice
- M. Insurance and Indemnity
- N. Respect for Privacy
- O. Compliance
The safety and wellbeing of the individual take precedence over the interests of science and society.
Every individual involved in the research process, including study management and general execution, are qualified by education, training and experience, or are otherwise deemed competent under the supervision of an appropriately qualified person, to perform their tasks.
Research projects are scientifically sound and are guided by ethical principles in all their aspects.
Patients, service users, and/or members of the public are involved in the design, management, conduct, dissemination, and application of research, unless otherwise justified.
Research is designed, reviewed, managed, and undertaken in a way that ensures scientific integrity, high quality outputs, and transparency.
The design and procedure are clearly described in both the research proposal, any ethics submission, and the protocol or manual of procedures. Where applicable, these documents should conform to a standard template and/or should contain any specified contents.
The researchers and sponsor familiarise themselves with any relevant legislation or regulatory requirements with regards to managing and conducting the project.
Any anticipated benefit for the individual participant and other present and future recipients of the health or social care in question is weighed against the foreseeable risks and inconveniences once they have been mitigated. A formal risk assessment is only expected where identified as essential. The risk:benefit ratio should be sufficiently described and considered as part of review processes such as REC review.
The research project is only commenced once all appropriate and required permissions and approvals are in place. This could include REC, MHRA, HRA, or ARSAC favourable opinions, and will depend on the research question being addressed / the processes being undertaken as part of the project.
To avoid waste, or the unnecessary duplication of projects, information about the research must be made publicly available prior to the commencement of the project. This does not include projects conducted for solely educational purposes.
The findings, whether positive or negative, must be made accessible, with adequate consent and privacy safeguards, in a timely manner after the project has finished, in compliance with any applicable regulatory standards. In addition, where appropriate, information about the findings of the research is available, in a suitable format and timely manner, to those who took part in it.
These requirements do not apply to projects conducted for solely educational purposes, or to early phase trials.
Research participants are afforded respect and autonomy, and the researchers must take account of their capacity to understand.
Where there is a difference between the research and the standard practice that they might otherwise experience, research participants are given information to understand the distinction and make an informed choice.
Where participants’ explicit consent is sought, it must be voluntary and informed. Where consent is refused or withdrawn, this must be done without reprisal.
Adequate provision is made for insurance or indemnity to cover any liabilities which may arise in relation to the design, management and conduct of the research project. Special provision is not expected unless existing arrangements (e.g. professional insurance, membership of NHS Litigation Authority schemes) provide inadequate cover.
All information collected for, or as part of, the research project is recorded, handled and stored appropriately and in such a way and for such time that it can be accurately reported, interpreted and verified, while the confidentiality of individual research participants remains appropriately protected.
Data and tissue collections are managed in a transparent way that demonstrates commitment to their appropriate use for research and appropriate protection of privacy.
Sanctions for non-compliance with these principles may include appropriate and proportionate administrative, contractual, or legal measures by funders, employers, relevant professional and statutory regulators, and other bodies.
In addition to the above, the following principles apply only to research where there is a change in the treatment, care, or other services applied to patients or participants for the purpose of research.
Principles that Apply to Interventional Health & Social Care Research
- P. Justified Intervention
- Q. Ongoing Provision of Treatment
- R. Integrity of the Care Record
- S. Duty of Care
The intended deviation from normal treatment, care, or other services is adequately supported and justified by existing evidence or scientific basis.
The research proposal or protocol and the participant information sheet explain the special arrangements, if any, after the research intervention period has ended. This could include either continuing or changing the treatment, care or other services that were introduced for the purposes of the research.
All information about any treatments, care or other services provided as part of the research project and their outcomes is recorded and stored in such a way, and for such time, that it can be understood by others involved in the participant’s care and accurately interpreted while the confidentiality of records of the participants remains protected.
The duty of care owed by health and social care providers continues to apply when their patients and service users take part in research. A relevant health or social care professional retains responsibility for the treatment, care, or other services provided to research participants, and for decisions about their treatment, care or other services. If a conflict arises between research and patient interests, the duty of care to the participant as a patient prevails.
Animals in Research
You should read and familiarise yourself with the Regulations outlined in the Animals (Scientific Procedures) Act 1986.
Some healthcare research projects will require non-human subject tests to be completed before the initial, first-in-human, trial. This usually begins with bench-top tests using synthetic organs or anatomical models and can involve the use of donated tissues or organs. Once the technology or system has been demonstrated to be functioning as intended, with a good degree of accuracy, then you may be able to move on to either animal or human clinical trials.
Research using animals is held to a very high ethical standard. Unlike humans, animals can’t consent to participate in research, so it’s vital that animal welfare is at the forefront of the project’s design. When you write your scheme of work for an animal project you must be considering the following:
The 3 R's
Avoid or replace the use of animals with a suitable alternative where possible. This could involve using organ analogues for bench-top testing, or designing robust models and tools that can simulate outcomes without using animals.
Minimise the number of animals used within a project. Appropriately and robustly designed projects will use the minimum possible number of animals whilst still obtaining valuable outcomes.
Minimise the suffering, distress, and lasting harm animals will experience. Adhere to the highest standards of animal welfare in practice and understand how animal distress can impact research outcomes.
Animal Welfare Considerations
- How many animals are you using?
- How will the animals be transported between facilities, and how will they be housed at facilities? Include details such as:
- Food and water provision
- Size of enclosures
- If the animals will be kept together, within sight of each other or separate, and why?
- Toys and enrichment tools
- Vehicles used for transport
- Enclosure and containment temperatures and humidity
- How often waste bedding or soiled items may be changed.
- What procedures will the animals be subject to, and how much distress do you reasonably expect the animal to experience, and how do you plan to adhere to the Principle of Refinement?
- What medications will be administered and how will they be administered? How much discomfort will the animal experience?
- For surgical procedures, how will the animals’ distress be minimised and managed before and during the procedure?
- What happens to the animals after the project?
- Euthanasia – The animal may be euthanised at the end of the procedures using approved methods such as anaesthetic overdose.
- Setting free – Animals procured from the wild may be set free, dependent on the provisions within the Animals (Scientific Procedures) Act 1986, and any other relevant legislation.
- Rehoming – Animals may be rehomed to a suitable facility or owner, dependent on the provisions within the Animals (Scientific Procedures) Act 1986, and any other relevant legislation.
The use of animals in research is, rightfully, heavily regulated. You may not conduct research using animals without the appropriate Site, Project, and Individual licenses in place. These licenses are applied for, and granted by, the Home Office.
You can find out further information about research using animals at Imperial here.
Download this Resource / Contact Us

You can download a copy of this resource here: Research Ethics at Imperial (PDF)
If you would like to contact us, or if there is a resource you would like to see, reach out to us using this form!
