Phylodynamic inference and model assessment with ABC
Oliver Ratmann et al. publish on using Approximate Bayesian Computations for phylodynamic inference and model assessment in PLoS Computional Biology.
Oliver Ratmann, Ge Donker, Adam Meijer and Christophe Fraser publish a methods paper on using Approximate Bayesian Computations for phylodynamic inference and model assessment in PLoS Computional Biology.
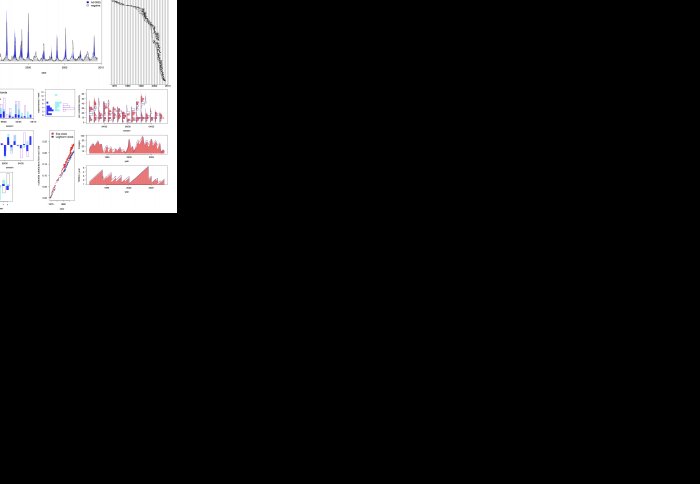
A key priority in infectious disease research is to understand the ecological and evolutionary drivers of viral diseases from data on disease incidence as well as viral genetic and antigenic variation. We propose using a simulation-based, Bayesian method known as Approximate Bayesian Computation (ABC) to fit and assess phylodynamic models that simulate pathogen evolution and ecology against summaries of these data. We illustrate the versatility of the method by analyzing two spatial models describing the phylodynamics of interpandemic human influenza virus subtype A(H3N2). The first model captures antigenic drift phenomenologically with continuously waning immunity, and the second epochal evolution model describes the replacement of major, relatively long-lived antigenic clusters. Combining features of long-term surveillance data from the Netherlands with features of influenza A (H3N2) hemagglutinin gene sequences sampled in northern Europe, key phylodynamic parameters can be estimated with ABC. Goodness-of-fit analyses reveal that the irregularity in interannual incidence and H3N2's ladder-like hemagglutinin phylogeny are quantitatively only reproduced under the epochal evolution model within a spatial context. However, the concomitant incidence dynamics result in a very large reproductive number and are not consistent with empirical estimates of H3N2's population level attack rate. These results demonstrate that the interactions between the evolutionary and ecological processes impose multiple quantitative constraints on the phylodynamic trajectories of influenza A(H3N2), so that sequence and surveillance data can be used synergistically. ABC, one of several data synthesis approaches, can easily interface a broad class of phylodynamic models with various types of data but requires careful calibration of the summaries and tolerance parameters.
Article text (excluding photos or graphics) © Imperial College London.
Photos and graphics subject to third party copyright used with permission or © Imperial College London.
Reporter
Oliver Ratmann
Department of Mathematics