Effect of Molecular Fluctuations on Hole Diffusion within Dye Monolayers
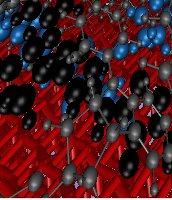
In dye sensitised solar cells, a dye monolayer (in gray) is sandwiched between a metal oxide surface (in red) and an electrolyte (purple).
Charge transfer is the fundamental process by which energy is transported through molecular materials.
Charge transfer is the fundamental process by which energy is transported through molecular materials. In dye sensitised solar cells for example, a current can be maintained in the dye monolayer by hole transfer from one molecule to another. The overall conductivity of the material can be derived from the kinetics of isolated charge transfer steps with multiscale modelling. As a result, adequately quantifying the rate of charge transfer remains an important issue in photovoltaics research.
In the high temperature limit, the non adiabatic Marcus’s theory allows the rate of charge exchange to be expressed as a function of the reorganisation energy and the electronic coupling. The latter strongly depends on the relative geometry of the charge donor with respect to the charge acceptor which makes molecular pair configuration an important parameter to determine. However, the monolayer is a highly disordered medium where the dyes are expected to adopt various configurations, fluctuating over time. A comprehensive simulation of these degrees of freedom is not trivial due to the localisation of the monolayer at the interface between a metal oxide substrate and an electrolyte.
A variety of models for charge transport have been derived to account for either very slow or very fast fluctuations in configurations (hence in electronic couplings), but mostly for organic crystals. In Chem. Mater. 2014, 26, 4731-4740, Vaissier et al. present methods to compute a series of surface anchored dye geometries as illustrated by the superimposition of potential conformations in the front page figure. The magnitude of the variation in electronic couplings was investigated and lead to the derivation of a multiscale framework for the simulation of hole diffusion in dye monolayers. The novelty of the model is the inclusion of the regime where the structural rearrangement occurs on a similar time scale as the charge transfer. The outcome is that large amplitude fluctuations of the tethered dyes at the microsecond timescale may enable charges to escape configurational traps. Therefore, the route towards highly conductive molecular materials might be to design fast-rearranging (rather than disorder-less) materials.

Illustration of the space spanned by the different conformations of a pair of surface anchored indolene dyes. The two figures are the front and side view of the superimposition of the frames, taken at regular interval, of a 9 ps Car-Parrinello molecular dynamics trajectory (red =surface, black = carbon and hydrogen, blue= sulfur and nitrogen).
Article text (excluding photos or graphics) © Imperial College London.
Photos and graphics subject to third party copyright used with permission or © Imperial College London.
Reporter
Veena Dhulipala
Registry